Projects
Metagenomic and genomic surveillance of antimicrobial resistance in hospital wastewater and computational prediction of efficacy of alternative non-antibiotic therapeutics (ATTACK-AMR)
The cooperative research project ATTACK-AMR involves teams from the US, Japan, Australia, Thailand, and the Philippines; and it aims to deliver alternative, non-antibiotic therapies to combat antimicrobial resistant (AMR) pathogens. AMR is one of the biggest challenges facing healthcare industries and is on a rapid rise as a result of the overuse of antibiotics. Replacing antibiotics with alternative products will delay the resistance and restore the activity of antibiotics that are no longer effective due to resistance.
In collaboration with our e-Asia JRP partners, the project aims to attack the problem of AMR in the Philippines on three fronts using next-generation sequencing technology and advanced machine-learning/AI tools. First, we will perform a resistome analysis of hospital waste-water metagenomes collected from 3 hospitals in Metro Manila. Similar metagenomic data will also be collected in partner countries. Bioinformatics analysis of the metagenomic data will provide insights into the abundance and diversity of AMR genes in our hospital wastewater and serve as a baseline measure for any future intervention studies. Next, we will collect, sequence, and analyze clinical isolates of P. aeruginosa and A. baumannii using available genome data. These isolates will be used and will be subjected to drug efflux pump inhibitors molecules identified by the Australian team. Finally, we will build system biology and machine-learning/AI models that can in-silico predict the efficacy of alternative therapies such as phage therapy and ecobiotics based on experimental data produced by the Thailand and US teams.
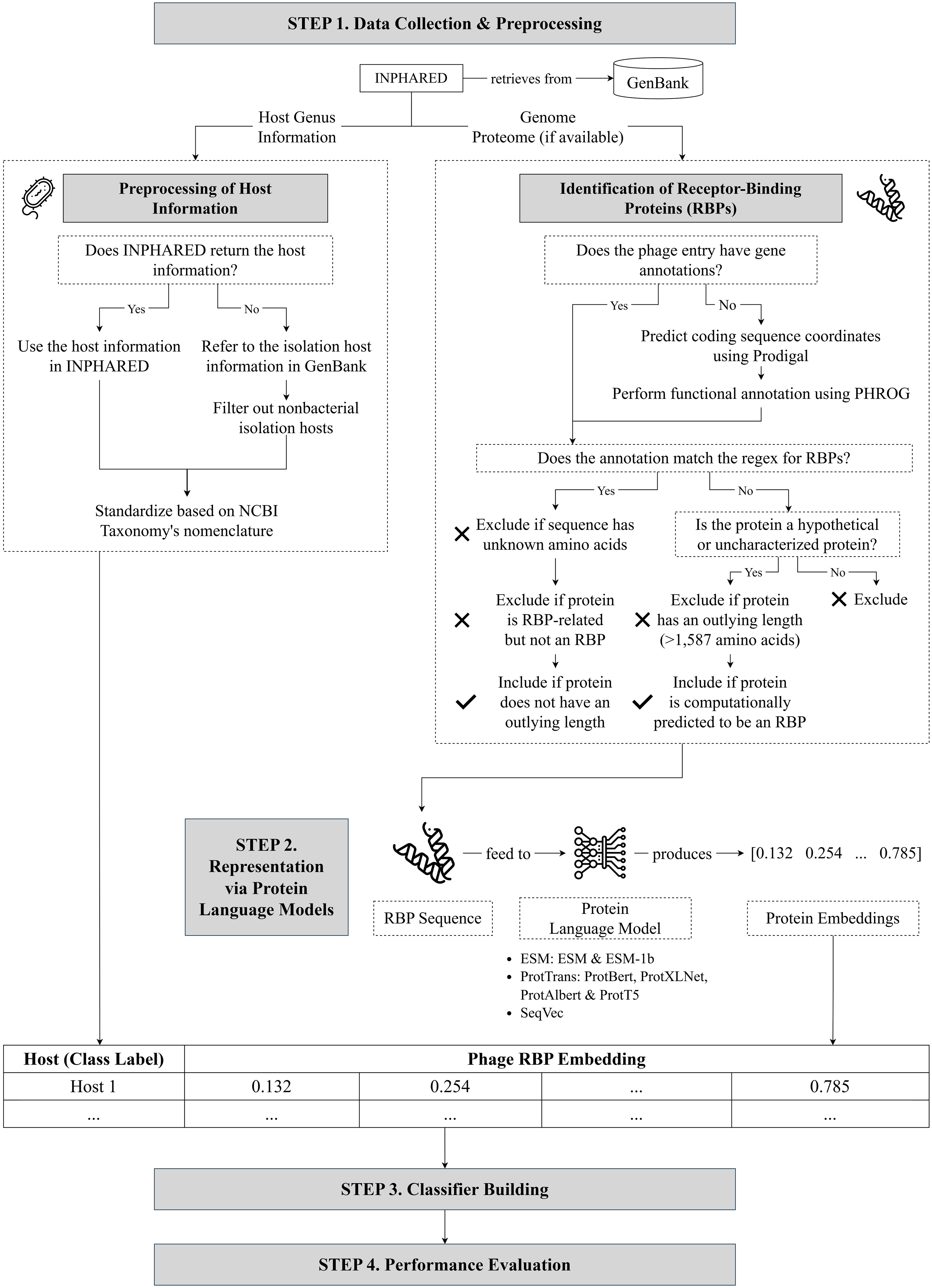
Protein embeddings improve phage-host interaction prediction
With the growing interest in using phages to combat antimicrobial resistance, computational methods for predicting phage-host interactions have been explored to help shortlist candidate phages. Most existing models consider entire proteomes and rely on manual feature engineering, which poses difficulty in selecting the most informative sequence properties to serve as input to the model. In this paper, we framed phage-host interaction prediction as a multiclass classification problem that takes as input the embeddings of a phage’s receptor-binding proteins, which are known to be the key machinery for host recognition, and predicts the host genus. We explored different protein language models to automatically encode these protein sequences into dense embeddings without the need for additional alignment or structural information. We show that the use of embeddings of receptor-binding proteins presents improvements over handcrafted genomic and protein sequence features. The highest performance was obtained using the transformer-based protein language model ProtT5, resulting in a 3% to 4% increase in weighted F1 and recall scores across different prediction confidence thresholds, compared to using selected handcrafted sequence features.
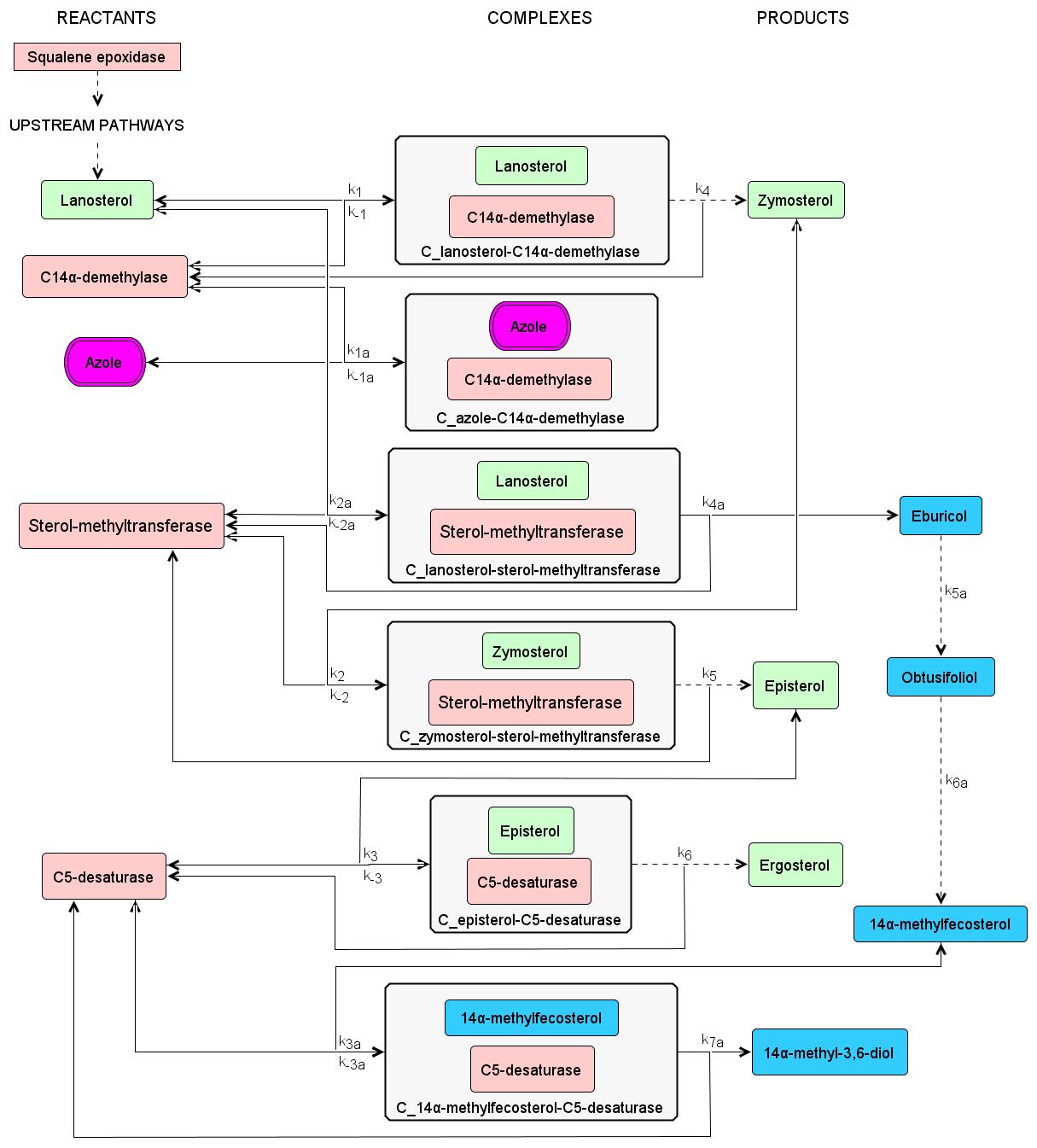
Systems biology approach of understanding azole resistance mechanisms in Candida albicans
Candida albicans is a commensal fungus pathogen commonly reported to cause candidiasis in immunocompromised patients. Noteworthy, it is reported as the most common fungal infection in the critical care setting. The current antifungal agent of choice is fluconazole, which is classified under the azole antifungals. It acts by inhibiting the ergosterol biosynthesis pathway, which is an essential component of the fungal cell membrane. However, in recent years, there has been a significant increase in fluconazole-resistant C. albicans as this has already been classified as a serious threat by the US Centers for Disease Control and Prevention. Hence, there is a need to investigate the azole resistance mechanisms of C. albicans and to find possible drug targets.
We aim to investigate the azole resistance mechanisms in Candida albicans using systems approach (mathematical and computational modeling) to identify important biomarkers for possible drug targets. The models aim to aid researchers narrow down the possible drug targets prior to doing costly and time-consuming experiments, and to serve as a cross-validation tool for experimental data.
Research Team
- Dr. Angelyn Lao
- Dr. Anish Shrestha
- Dr. Maria Luisa Enriquez
- Dr. Llewelyn Moron
- Dr. Evelina Lagamayo
- Dr. Edilberto Manahan
- Ms. Sherill Tesalona
- Mr. Joemarie Malana
Research/Student Assistants
- Robert D. Unciano , Project Assistant
- Jiaan Carlo E. Santos , Research Assistant
- Paul K. Yu
- Daphne Janelyn L. Go
- Mark Edward M. Gonzales
Implementing Agency
![]()
Co-implementing Agencies
![]()
![]()
![]()
![]()
Cooperating Agencies
![]()
![]()
![]()
Funding Agencies
![]()
![]()
Partners
![]()